
Identifying failed DNA sequencing reactions
- The basecall has only five N bases.
- The trace chromatogram has noisy or “messy” sequence peaks with low quality scores. The trace can have false peaks that look real and have high quality scores, especially when the trace has been base called using the KB Basecaller™.
- Little or no signal in the raw data channels except for leftover dye peaks.
- The peak heights in the raw channel are below 100.
- The trace sequence does not match either the expected sequence, or any other sequences in GenBank.
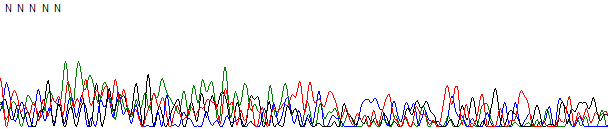
Figure 1. Failed reaction.
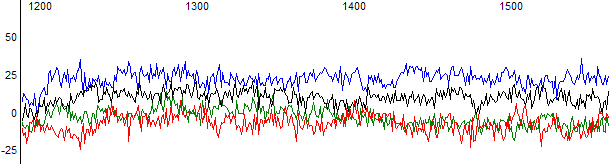
Figure 2. Raw data channel of a failed reaction.
Figure 1 shows an example of a failed reaction trace. In this example the trace has been basecalled by KB which provided five N bases and very noisy peaks. In the raw data channel (Figure 2) there is no obvious peaks and the signal is at the sequencer noise level.
Causes of failed DNA sequencing reactions
- Poor quality DNA. Very common when sequencing plasmid DNA templates. A common cause is not removing all the SDS detergent from the miniprep.
- Loss of the sequencing reaction products during clean up. This is a particular problem when using ethanol precipitation clean up protocols.
- Too much or too little template DNA. Excess template DNA can kill the sequencing reaction and not adding enough can prevent enough labelled fragments being generated.
- The wrong primer was used. This is more common than you might think!
- Bad water. The water used contains a sequencing inhibitor. This can occur if there is bacterial growth in the Milli-Q hose.
- Degraded or failed synthesis primer. Oligonucleotide synthesis is chemically complex and primer synthesis failures are fairly common. The primers once diluted down to working concentrations are susceptible to freeze-thaw degradation or DNase contamination.
- Dead sequencing chemistry. Can occur if the BigDye™ chemistry is stored under the wrong conditions or it is freeze-thawed too many times. Either the Taq DNA polymerase or dye labeled nucleotides can degrade.
- Blocked capillary. If this occurs, then every trace using a particular capillary fails. This can be identified by tracking trace quality on a trace by trace basis.
Solving DNA sequencing reaction failures
- Poor quality DNA. The best way of avoiding this problem is to not sequence plasmid DNA and sequence a PCR amplified fragment of the plasmid insert. If this is not possible then it is recommended that a plasmid miniprep kit is used. One tip is to perform a final ethanol precipitation on the kit purified plasmid DNA. This often solves problems with the quality of the template (provided you don’t lose the DNA pellet).
- Loss of sequencing reaction during the clean up step. This can be avoided by not using an ethanol precipitation protocol to clean up the sequencing reaction. There are a number of kits that work very well, unfortunately they can be expensive. One tip for avoiding the loss of the reaction DNA pellet when using an ethanol precipitation protocol is to add 1 µl of a 20 mg/ml solution of glycogen (Sigma G-1508) to the sequencing reaction before adding the ethanol. This helps make the pellet visible and the glycogen does not seem to interfere with the injection of the sequencing DNA fragments onto the DNA sequencer capillaries.
- Too much or too little template DNA was used. This can be avoided by checking the concentration of the template on an agarose gel before sequencing. This will also allow you to see the purity of the template DNA and if there is a significant amount of contaminating genomic DNA or RNA present. Do not rely on a spectrophotometer reading to calculate the template concentration.
- Wrong primer. This is simple to solve, but can be difficult to detect. Check the sequence of the primer and template to make sure that the primer binding site is present. This can be a particular problem with some “universal” forward and reverse primer sequences which do not work with some common plasmids. Do not trust other people’s working stock solutions (especially those of your unreliable lab colleagues!) and make your own. It might take 5 minutes longer, but it will save you a lot of future headaches.
- Bad water. Inhibitors can contaminate lab water stocks that can kill DNA sequencing reactions. If you think this may be a problem, then throw out your water and use a fresh stock – remember water is cheap!
- Degraded primer. Don’t use old diluted primer stocks. Store the primers in 10 mM Tris / 0.1 mM EDTA (pH 8.5) rather than water. Don’t use other people’s primer stocks. If you have any doubt about the oligonucleotide primer quality, throw it out and make up a fresh working solution from stock.
- Failed oligonucleotide synthesis. If you suspect that the primer is poor quality, either have it resynthesized or check the primer in a polymerase chain reaction (PCR). Alternatively, if you have a control template that you know should work with the primer, then this can be a good way of identifying primer problems.
- Dead sequencing chemistry. This is a relatively rare problem, however, if a batch of BigDye chemistry has not been used for some time, or there is any doubt about how it has been stored, then it is advisable to perform a control sequencing reaction before undertaking a large number of experimental reactions. Many problems with dead chemistry can be prevented by storing the BigDye chemistry in small aliquots and avoiding repeated freeze/thaw cycles.
- Blocked capillary. Can be identified by tracking trace quality on a trace by trace basis. While this can be done manually, we recommend using our QualTrace III software. If you suspect that a sequencer capillary is blocked, then inform the sequencer operator.
For more information on automated QC tracking of sequencing traces please visit the QualTrace III DNA sequencing analysis software page.
Return to the main DNA sequencing troubleshooting page.