In most scenarios the usage of PeakTrace will lead to an increasing the number of high quality bases in the basecall. There are however a few factors that can limit PeakTrace’s ability to improve a .ab1 trace file, such as:
- Length of read. Shorter reads will already have been basecalled near perfectly by KB so any improvement will be minimal.
- Raw channel signal. If the signal is at noise level (just detector noise) it may not be possible to improve over KB.
- Failed reactions. If a sequencing reaction has failed (no data to process) not even PeakTrace can salvage it.
- Mixed trace signal. DNA contamination (two or more templates or priming sites) cannot be removed through PeakTrace processing.
If PeakTrace encounters a trace it can’t improve there are two ways to deal with the original file. The first involves just copying the .ab1 file to the output folder with no attempts at improving the file, or the appearance of the trace can be improved through KB fallback processing with the Clean Baseline, Extra Baseline and Extra Normalization options.
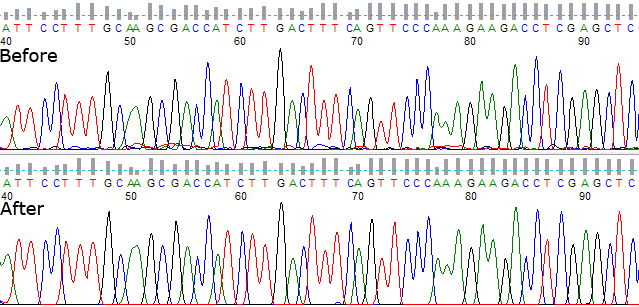
If PeakTrace can’t improve the basecall sequence but you want to improve the original input file select PeakTrace/KB basecaller.
Figure 1. PeakTrace basecalling with KB fallback.
If PeakTrace can’t improve the basecall sequence and you do not wish to improve the original file select PeakTrace basecaller.
Figure 2. PeakTrace basecalling with no fallback.
Be aware that improving a file through the use of the Clean Baseline, Extra Baseline or Extra Normalization options will use a processing unit even if PeakTrace hasn’t basecalled the file.